

Actualización en Diagnóstico y Manejo Multidisciplinario
1. ¿Qué es el Síndrome de Ehlers-Danlos?
Los Síndromes de Ehlers-Danlos (SED) conforman un grupo de enfermedades hereditarias del tejido conectivo. Su origen radica en alteraciones en la síntesis, estructura o procesamiento del colágeno y las proteínas de la matriz extracelular: el «andamiaje» que mantiene unidas las estructuras del cuerpo.
La clasificación internacional de 2017 reconoce 13 subtipos con bases moleculares y manifestaciones clínicas distintas. El más frecuente es el subtipo hipermóvil (hSED), que representa aproximadamente el 90 % de los casos. Su particularidad es que, hasta la fecha, carece de una causa genética identificada: el diagnóstico es exclusivamente clínico.

Existe además el Trastorno del Espectro de Hipermovilidad (TEH), una entidad clínicamente similar al hSED pero que no cumple la totalidad de los criterios diagnósticos. Ambas condiciones forman parte de un continuum: en un extremo, la hiperlaxitud articular asintomática; en el otro, el hSED con carga de enfermedad severa y compromiso multisistémico.
La comprensión de este espectro ha evolucionado profundamente en la última década, transitando desde un enfoque puramente reumatológico hacia un modelo biopsicosocial que reconoce la complejidad de estos pacientes.
2. ¿Cómo se diagnostica?
El diagnóstico de hSED se basa en los criterios publicados en 2017 por Malfait et al. en el American Journal of Medical Genetics. Requiere tres condiciones fundamentales:
- Hipermovilidad articular generalizada, evaluada con la Escala de Beighton.
- Presencia de dos o más características secundarias (manifestaciones sistémicas, historia familiar o complicaciones musculoesqueléticas).
- Ausencia de hallazgos que sugieran otro subtipo de SED u otro trastorno del tejido conectivo.
2.1 La Escala de Beighton: evaluando la hiperlaxitud
Esta escala puntúa cinco maniobras articulares sobre un máximo de 9 puntos. Se considera positiva para hiperlaxitud generalizada si se alcanzan los siguientes umbrales:
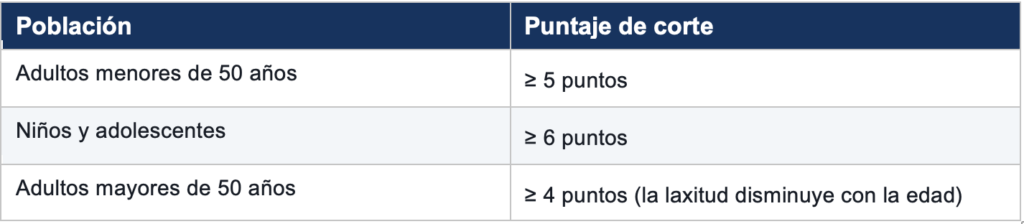
Las 5 maniobras evaluadas
- Hiperextensión del 5° dedo (meñique) ≥ 90° con el antebrazo apoyado — mano derecha e izquierda (2 puntos).
- Oposición pasiva del pulgar al antebrazo flexionando la muñeca — derecha e izquierda (2 puntos).
- Hiperextensión del codo > 10° — derecho e izquierdo (2 puntos).
- Hiperextensión de rodilla > 10° (genu recurvatum) — derecha e izquierda (2 puntos).
- Flexión del tronco hacia adelante con rodillas extendidas, palmas completamente apoyadas en el suelo (1 punto).

2.2 Manifestaciones secundarias que orientan el diagnóstico
Las características secundarias se agrupan en tres criterios:
Criterio A — Manifestaciones sistémicas (al menos 5 de 12)
- Piel aterciopelada o con hiperextensibilidad leve.
- Estrías sin causa aparente.
- Pápulas piezogénicas en talones, hernias recurrentes.
- Cicatrices atróficas, prolapso de órganos pélvicos.
- Insuficiencia venosa crónica.
- Habitus marfanoide, aracnodactilia, pie plano.
- Subluxaciones o luxaciones articulares.
Criterio B — Historia familiar
- Pariente de primer grado con diagnóstico confirmado de hSED.
Criterio C — Complicaciones musculoesqueléticas
- Dolor crónico generalizado por más de 3 meses.
- Inestabilidad articular recurrente o luxaciones sin traumatismo.
- Dolor articular crónico a pesar de radiografías normales.
2.3 Diagnóstico diferencial: lo que hay que descartar
Antes de confirmar el diagnóstico de hSED es indispensable descartar otros subtipos de SED y enfermedades del tejido conectivo. Algunos requieren confirmación genética urgente:
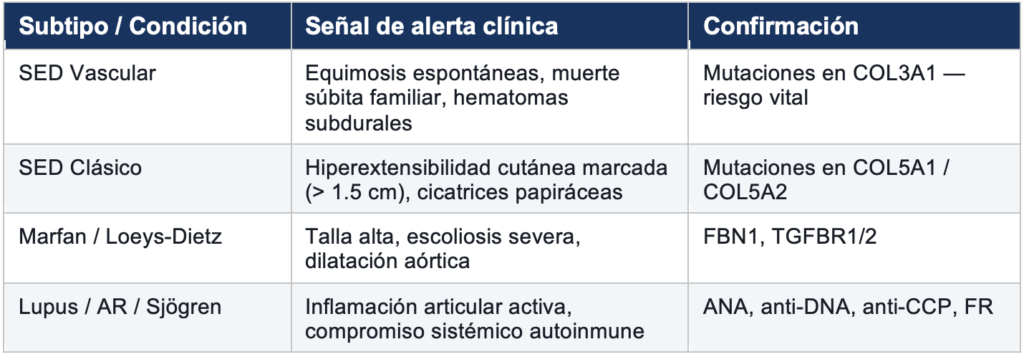
2.4 Estudio de laboratorio
No existe un examen de laboratorio que confirme el hSED. Los exámenes sirven para descartar otras enfermedades:
Screening general
- Hemograma completo, VHS y PCR (descartar inflamación activa).
- Perfil bioquímico, función renal y hepática.
- CPK (descartar miopatía), TSH y T4 libre (descartar hipotiroidismo).
- Vitamina D, ferritina y vitamina B12 (contribuyentes frecuentes a fatiga y dolor musculoesquelético).
Panel autoinmune (cuando hay sospecha clínica)
- ANA, anti-DNA, ENA: descartar lupus y síndrome de Sjögren.
- Factor reumatoide y anti-CCP: descartar artritis reumatoide.
- Complemento C3 y C4 si se sospecha enfermedad autoinmune.
3. Manejo del Dolor Crónico
El dolor crónico es la manifestación más discapacitante del hSED: afecta al 90-100 % de los pacientes con intensidad moderada a severa. Su origen es multifactorial, lo que exige un enfoque terapéutico igualmente complejo:
- Dolor nociceptivo por microtraumatismos articulares repetidos.
- Dolor neuropático por atrapamiento nervioso y neuropatía de fibras pequeñas.
- Dolor nociplástico por sensibilización central del sistema nervioso.
- Componente miofascial secundario a patrones compensatorios.
3.1 Neuromoduladores: el pilar del tratamiento farmacológico
Los neuromoduladores son los fármacos con mayor evidencia para el dolor en hSED. Actúan modulando la transmisión del dolor en el sistema nervioso central y periférico.
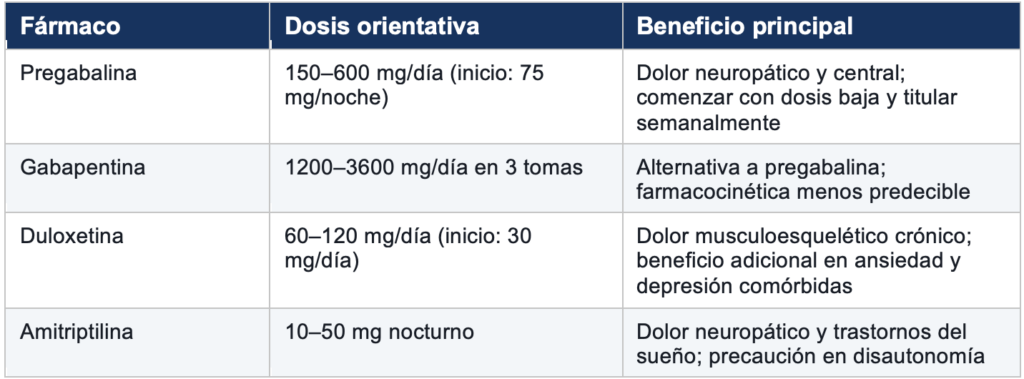
3.2 Analgésicos complementarios
Los analgésicos simples tienen un rol de apoyo. El paracetamol (1 g cada 6-8 horas) puede ser útil para el dolor leve a moderado con buena seguridad. Los antiinflamatorios no esteroidales (AINEs) deben usarse con precaución: el celecoxib (200 mg/día) ofrece menor toxicidad gastrointestinal cuando es necesaria la cobertura antiinflamatoria.

3.3 Terapias emergentes
En el contexto de dolor refractario y bajo supervisión especializada, pueden considerarse opciones como cannabinoides (con evidencia preliminar en dolor neuropático), ketamina en dosis subanestésicas (para sensibilización central severa) y lidocaína transdérmica para dolor neuropático localizado.
4. Rehabilitación: el Tratamiento con Mayor Evidencia
La rehabilitación es la intervención con el nivel de evidencia más alto en hSED y TEH, superior a cualquier tratamiento farmacológico aislado. Su objetivo central es optimizar el control neuromuscular y la estabilidad articular dinámica, compensando la laxitud ligamentaria intrínseca.
El enfoque moderno es radicalmente distinto al histórico: en lugar de énfasis en estiramiento, se prioriza fortalecimiento, reeducación propioceptiva y control motor.
4.1 Principios del ejercicio terapéutico
Los protocolos de ejercicio en hSED deben adaptarse a la biomecánica particular de estos pacientes:
- Fortalecimiento muscular en rangos medios de movimiento, evitando posiciones de hiperextensión.
- Ejercicios isométricos e isotónicos en cadena cinética cerrada, preferibles a movimientos balísticos.
- Series de 12-15 repeticiones con énfasis en contracción excéntrica controlada.
- Progresión gradual desde carga mínima con alto control motor hacia cargas moderadas.
- El estiramiento pasivo agresivo debe evitarse: perpetúa la inestabilidad y aumenta el riesgo de subluxaciones.
4.2 Reeducación propioceptiva
La reeducación propioceptiva es un pilar fundamental. En hSED existe un procesamiento sensoriomotor deficiente que contribuye a la inestabilidad articular. Se trabaja con:
- Ejercicios en superficies inestables y balance unipodal.
- Perturbaciones controladas e integración de feedback visual y táctil.
- Sesiones diarias de 10-15 minutos de entrenamiento propioceptivo específico.
Estudios prospectivos muestran reducción significativa de subluxaciones y mejoría funcional con este abordaje.
4.3 Protocolos estructurados
El Muldowney Protocol es el protocolo de rehabilitación más aceptado en la comunidad de hSED. Se extiende durante 12 semanas y progresa sistemáticamente desde la estabilización central (core) hacia las extremidades, enfatizando alineación postural y patrones de movimiento funcional. Los estudios observacionales muestran mejoría sostenida en dolor, función y calidad de vida a 6-12 meses en pacientes adherentes.
4.4 Modalidades complementarias
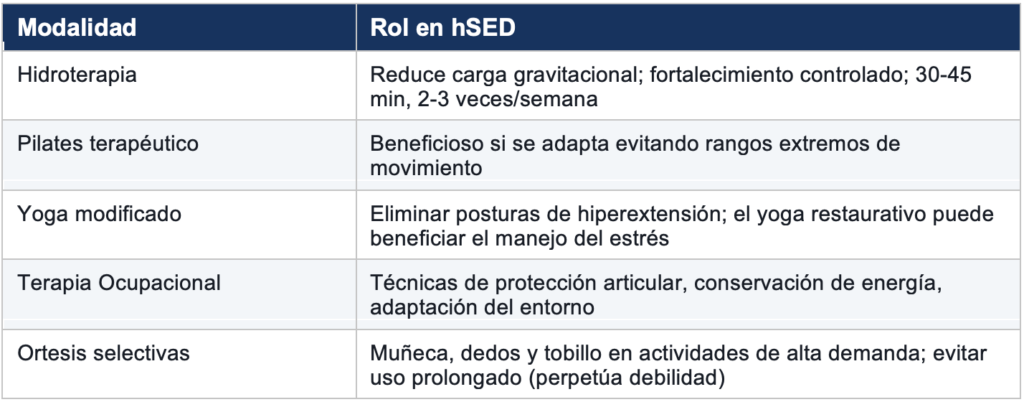
4.5 Actividad física a largo plazo
La progresión hacia actividad física regular es el objetivo final. Se recomiendan actividades de bajo impacto: ciclismo, elíptica, caminata en terreno plano, natación (evitando brazada de mariposa). El entrenamiento de fuerza supervisado, 2-3 veces por semana, debe ser un componente permanente del manejo.

5. Manifestaciones Asociadas y Comorbilidades
El hSED es mucho más que una enfermedad articular. Las manifestaciones extrararticulares frecuentemente dominan el cuadro clínico y determinan la calidad de vida del paciente. El manejo integral debe abordarlas en paralelo.
5.1 Disautonomía y POTS
El síndrome de taquicardia postural ortostática (POTS) afecta al 25-50 % de los pacientes con hSED. Se manifiesta con:
- Taquicardia excesiva al ponerse de pie (aumento ≥ 30 lpm o frecuencia ≥ 120 lpm).
- Mareo, presíncope, fatiga, intolerancia al ejercicio.
- Disfunción cognitiva («brain fog»).
El tratamiento es escalonado: primero medidas no farmacológicas (incremento de sal 10-12 g/día, hidratación 2-3 L/día, medias de compresión 30-40 mmHg, ejercicio en posición horizontal progresando a bipedestación). Si no es suficiente, se puede recurrir a fludrocortisona, midodrina, betabloqueantes en dosis bajas, piridostigmina o ivabradina.
5.2 Fatiga crónica
Afecta al 80-90 % de los pacientes con hSED y frecuentemente cumple criterios de síndrome de fatiga crónica. Su origen es multifactorial: disfunción mitocondrial, disautonomía, trastornos del sueño, dolor crónico y desacondicionamiento. El manejo requiere abordar cada factor contribuyente de forma individual y coordinada.
5.3 Activación mastocitaria
Una comorbilidad recientemente reconocida, con prevalencia estimada del 20-40 % en hSED. Los síntomas incluyen flushing, urticaria, dolor abdominal, diarrea y episodios de anafilaxia. El tratamiento combina antihistamínicos H1 y H2, cromoglicato sódico, ketotifeno y montelukast, junto con la identificación y evitación de desencadenantes.
5.4 Compromiso gastrointestinal
Incluye dispepsia funcional, síndrome de intestino irritable, náusea crónica, disfagia y gastroparesia. El manejo combina modificaciones dietéticas (dieta baja en FODMAPs, comidas pequeñas y frecuentes), procinéticos y tratamiento de sobrecrecimiento bacteriano si se documenta.
5.5 Comorbilidades psiquiátricas
La prevalencia de trastornos del ánimo y de la conducta en hSED supera ampliamente a la población general:
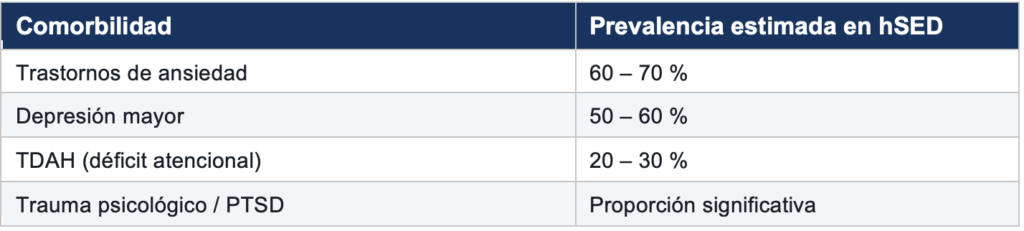
La relación es bidireccional: el dolor crónico y la limitación funcional exacerban los síntomas psiquiátricos, mientras que la disfunción autonómica y el procesamiento alterado de amenaza pueden compartir bases neurobiológicas. La psicoterapia (TCC, ACT, EMDR) y el apoyo farmacológico son componentes esenciales del tratamiento integral.

6. Conclusiones
El síndrome de Ehlers-Danlos hipermóvil y el trastorno del espectro de hipermovilidad son condiciones complejas cuyo manejo ha evolucionado hacia un modelo multidisciplinario centrado en la rehabilitación. Algunos puntos fundamentales a retener:
- El diagnóstico es clínico. Se basa en los criterios de 2017 y en la exclusión cuidadosa de otros trastornos del tejido conectivo, especialmente el SED vascular por su riesgo vital.
- El retraso diagnóstico es un problema real y evitable. Reconocer el espectro clínico desde la consulta inicial es fundamental.
- La rehabilitación es el tratamiento más efectivo. El fortalecimiento muscular y la reeducación propioceptiva superan en evidencia a cualquier intervención farmacológica aislada.
- El manejo farmacológico del dolor se basa en neuromoduladores. Los opioides deben evitarse en el contexto crónico.
- Las comorbilidades son parte del cuadro. Disautonomía, fatiga, activación mastocitaria y trastornos psiquiátricos deben abordarse en forma coordinada.
- El cambio de estilo de vida es permanente. Los beneficios de la rehabilitación se sostienen solo con adherencia a largo plazo.
Actualmente no existen terapias modificadoras de la enfermedad. La investigación futura debe priorizar la identificación de biomarcadores moleculares en hSED, ensayos clínicos controlados y modelos de atención integrada. Mientras tanto, la intervención rehabilitadora temprana y sostenida es la herramienta más poderosa para prevenir la progresión hacia la discapacidad.
Bibliografía Seleccionada
Referencias basadas en evidencia 2009–2024
1. Malfait F, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C. 2017;175(1):8-26.
2. Castori M, et al. A framework for the classification of joint hypermobility and related conditions. Am J Med Genet C. 2017;175(1):148-157.
3. Engelbert RH, et al. The evidence-based rationale for physical therapy treatment in hEDS. Am J Med Genet C. 2017;175(1):158-167.
4. Palmer S, et al. The effectiveness of therapeutic exercise for joint hypermobility syndrome: a systematic review. Physiotherapy. 2014;100(3):220-227.
5. Chopra P, et al. Pain management in the Ehlers-Danlos syndromes. Am J Med Genet C. 2017;175(1):212-219.
6. De Wandele I, et al. Orthostatic intolerance and fatigue in the hypermobility type of EDS. Rheumatology. 2016;55(8):1412-1420.
7. Seneviratne SL, et al. Mast cell disorders in Ehlers-Danlos syndrome. Am J Med Genet C. 2017;175(1):226-236.
8. Bulbena A, et al. Psychiatric and psychological aspects in the Ehlers-Danlos syndromes. Am J Med Genet C. 2017;175(1):237-245.
9. Fikree A, et al. Gastrointestinal involvement in the Ehlers-Danlos syndromes. Am J Med Genet C. 2017;175(1):181-187.
10. Demmler JC, et al. Diagnosed prevalence of EDS and HSD in Wales, UK: a national cohort study. BMJ Open. 2019;9(11):e031365.
11. Scheper MC, et al. Disability in adolescents and adults with hypermobility-related disorders: a meta-analysis. Arch Phys Med Rehabil. 2016;97(12):2174-2187.
12. Voermans NC, et al. Pain in Ehlers-Danlos syndrome is common, severe, and associated with functional impairment. J Pain Symptom Manage. 2010;40(3):370-378.
13. Lam CY, et al. Rome IV functional GI disorders in hypermobility spectrum disorders. Clin Gastroenterol Hepatol. 2021;19(2):277-287.
14. Rowe PC, et al. Orthostatic intolerance and chronic fatigue syndrome associated with Ehlers-Danlos syndrome. J Pediatr. 1999;135(4):494-499.
15. Henderson FC Sr, et al. Neurological and spinal manifestations of the Ehlers-Danlos syndromes. Am J Med Genet C. 2017;175(1):195-211.